
✅ 本文基于FDA公开法规与检查程序手册整理,聚焦2026年QMSR生效背景下的检查要点与企业实操准备。
? 写在前面:为什么这篇文章值得读
FDA对医疗器械制造商的现场检查,是很多企业进入美国市场的“必过关卡”。但长期以来,行业讨论往往集中在“483表开了几条”这样的结果指标上,却很少有人系统拆解检查员背后的工作逻辑。
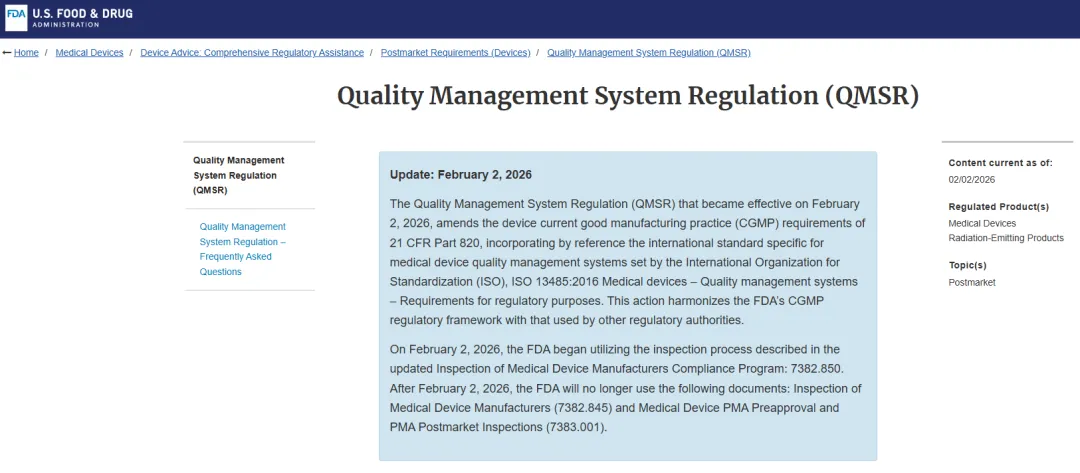
? 2026年2月,FDA正式完成了一次影响深远的法规切换:质量体系法规(QSR)被质量管理体系法规(QMSR)取代,同时启用全新的检查合规程序手册CP 7382.850。这意味着,检查员的关注点、取证方式、判断标准都与过去有了实质性不同。
? 本文试图回答三个问题:
FDA检查员在进场之前、现场之中、离场之后,分别做什么?
哪些环节最容易成为检查突破口?
企业如何提前构建一套能“自我证明”的质量体系?
? 第一部分|检查从来不是从进门开始的 FDA检查员的准备工作,远比大多数企业想象的更早、更深。
? 在确定检查计划后,检查员会通过FDA内部数据库和公开渠道,提前收集以下信息:
企业的注册信息和产品列名状态
510(k)或PMA的批准记录及任何补充文件
UDI编码及其在GUDID数据库中的提交完整性
过往检查报告(尤其是483表和警告信)
医疗器械报告(MDR)数据库中的不良事件记录
召回、纠正和移除的相关信息
? 此外,FDA自2025年6月起在全中心部署了Elsa生成式AI平台,该平台可自动分析不良事件趋势、合规异常和历史检查结果,帮助检查员在出发前就锁定高风险领域。
⚠️ 对企业意味着:如果不提前自查注册列名有效性、UDI数据一致性、MDR提交记录等基础信息,现场检查时连“解释”的机会都很有限。
? 第二部分|新检查框架下的核心关注域 2.1 从QSIT到QMSR:检查逻辑的深层变化
? 在2026年2月之前,FDA长期使用“质量体系检查技术”(QSIT)作为检查方法,该方法将质量体系拆分为管理控制、设计控制、生产与过程控制、纠正预防措施(CAPA)等若干子系统,自上而下逐项核对。
? 随着QMSR的生效和CP 7382.850的启用,这一方法被全面替换。新程序不再沿用QSIT的子系统切割方式,而是要求检查员基于风险和数据驱动的思路,跨系统、跨流程地评估整个质量管理体系的有效性。QMSR正式纳入了ISO 13485:2016作为核心要求,同时引用ISO 9000:2015统一术语。
? 检查的核心问题从“你有没有这个文件”变成了“你的体系在实际运行中是否保持一致、能否形成闭环”。
2.2 内部审计与管理评审:一个容易被低估的变化
? 在过去二十多年的QSIT框架下,FDA检查员通常不会要求查看内部审计报告、管理评审记录或详细的供应商审计报告。这并非法律明文禁止,而是基于FDA内部政策形成的长期惯例——检查员可以确认企业“已完成内审/管理评审”的证据,但不能审阅报告的具体内容。
⚡ QMSR彻底改变了这一局面。通过纳入ISO 13485:2016的第8.2.2条(内部审计)和第5.6条(管理评审),FDA首次从法规层面明确授予检查员审查以下文件的权限:
? 内部审计的完整报告及支持性文档
? 管理评审会议的记录与决议
? 供应商审计的记录
? 相关培训文档
? 这意味着,企业不能再将内部审计和管理评审视为“内部私密文件”而拒绝提供。检查员还会将审计发现与本次检查结果进行比对,以验证内部审计程序是否真正有效。
2.3 投诉、MDR与CAPA的联动审查
? 上市后监督是FDA当前的重点关注领域。根据公开数据,2023年10月至2025年9月期间,FDA共开展了约4,400次医疗器械检查,其中38%被归类为上市后监督活动。
? 检查员在检查前就会调取企业的MDR数据。现场检查时,他们通常采用“样本追溯法”:随机选取几条投诉记录,然后逐项追问——
该投诉是否经过了充分调查?
调查结论是否触发了MDR判断?是否按规定时限提交了MDR?
如果启动了CAPA,纠正措施是否延伸到设计控制、生产过程或风险管理?
变更是否经过了验证?是否更新了相关文件?
❌ 未按规定提交MDR的行为,将被直接列入483表。 更重要的是,这条追溯路径实质上是在检验:企业的质量体系是否具备真正的闭环能力,而不仅仅是处理一个个孤立的客诉。
2.4 UDI与GUDID:数据一致性正成为执法重点
?️ UDI法规的实施采取分阶段合规日期的策略,不同类别的器械截止时间不同。对于已经到期的类别,UDI是强制性要求。
⚠️ FDA的执法行动正越来越多地聚焦于数据质量。现场检查中,检查员会直接核实物上的UDI编码与GUDID数据库中的记录是否一致——包括器械标识符(DI)、包装层级标识、生产标识符(如适用)以及当前状态。
? 一个UDI数据差异,足以构成483观察项。 此前已有企业因UDI标签不合规或GUDID信息不完整而收到警告信。
2.5 483表:从“恐惧”到“理性应对”
? FDA在检查结束时如果发现重大不符合项,会出具FDA 483表(检查观察报告)。这是一个事实陈述,而不是最终处罚。
⏱️ 企业在收到483表后,通常有15个工作日提交书面回应。部分检查员也允许企业在现场对483表的观察项进行批注,例如标注“已纠正并验证”“承诺纠正”等。但需要明确的是,现场批注并不是企业的强制性权利——是否接受、如何接受,取决于检查员的现场判断。
?️ 如果能在检查员离场前完成某些问题的整改并获得认可,确实有助于降低后续升级为警告信的风险。但最终是否发出警告信,取决于企业提交的正式整改回复是否能从体系层面解决根本问题。
? 值得注意的是,FDA的警告信数量正在上升。截至2025年9月初,针对医疗器械质量体系违规发出的警告信已达19封,超过了2024年同期总和。
?️ 第三部分|企业可以提前构建的四项能力 ✅ 能力一:把“证据链思维”嵌入日常运行
不要等到检查通知来了再去“补记录”。真正有效的准备,是在日常运行中就让每一条客诉、每一次变更、每一个CAPA都能够被追溯:
客诉是否经过了闭环调查?
是否触发了MDR判断?判断过程是否有记录?
CAPA的原因分析是否反推到设计或过程?
验证结果是否及时归档?
? 这套证据链不需要刻意“为检查而做”,但它必须是完整、可查阅、经得起抽样的。
✅ 能力二:以CP 7382.850为蓝本开展模拟演练
? 企业可以按照新合规程序手册的逻辑,定期组织内部模拟检查。模拟不应只停留在“看文件”,而要覆盖:
文件调取的速度与准确性
现场陪检人员的应答规范
跨部门信息协同的顺畅度
对检查员追问的应对能力
✅ 能力三:将内部审计和管理评审升级为“合规预检”
? 既然这些文件已经进入可检查范围,企业就不应再将其视为内部保密文件而敷衍了事。管理评审应当真实反映质量体系的运行状况,内部审计应当覆盖关键风险区域,审计发现与整改措施必须形成闭环记录。
✏️ 一个实用的做法:在每次管理评审或内部审计结束后,问自己一个问题——“如果检查员现在看到这份记录,他会认为我们的体系是有效的,还是有明显漏洞?”
✅ 能力四:提前完成UDI与GUDID的全量核查
? 建议企业对所有已到合规日期的产品,逐一比对:
实物标签上的UDI编码
包装层级的UDI编码
GUDID数据库中对应的记录
⚙️ 确保三端完全一致。同时,确认海外代理人具备足够的专业能力,能够在必要时协助FDA安排检查、传递信息,而不仅仅是一个“信箱”角色。

? 写在最后
FDA的检查方法从QSIT转向CP 7382.850,不是一次简单的版本更新,而是一次监管思维的系统性升级——从“查文件”走向“查系统”,从“看单点”走向“看闭环”。
? 能够从容应对这种检查的企业,不是那些临时抱佛脚、突击补记录的团队,而是那些在日常运营中已经把质量证据链做得清晰、完整、可追溯的团队。
? 合规从来不是一个部门的战斗,更不是一次检查的输赢。它是企业进入美国市场最基本的入场券,也是长期竞争中最扎实的护城河。
免责声明:本文内容仅供信息参考。FDA法规和检查要求可能随时更新,具体合规事宜请以FDA官方最新发布为准。
信息来源:网络新闻
排版整理:金飞鹰药械
✅ 本文基于FDA公开法规与检查程序手册整理,聚焦2026年QMSR生效背景下的检查要点与企业实操准备。
? 写在前面:为什么这篇文章值得读
FDA对医疗器械制造商的现场检查,是很多企业进入美国市场的“必过关卡”。但长期以来,行业讨论往往集中在“483表开了几条”这样的结果指标上,却很少有人系统拆解检查员背后的工作逻辑。
? 2026年2月,FDA正式完成了一次影响深远的法规切换:质量体系法规(QSR)被质量管理体系法规(QMSR)取代,同时启用全新的检查合规程序手册CP 7382.850。这意味着,检查员的关注点、取证方式、判断标准都与过去有了实质性不同。
? 本文试图回答三个问题:
FDA检查员在进场之前、现场之中、离场之后,分别做什么?
哪些环节最容易成为检查突破口?
企业如何提前构建一套能“自我证明”的质量体系?
? 第一部分|检查从来不是从进门开始的 FDA检查员的准备工作,远比大多数企业想象的更早、更深。
? 在确定检查计划后,检查员会通过FDA内部数据库和公开渠道,提前收集以下信息:
企业的注册信息和产品列名状态
510(k)或PMA的批准记录及任何补充文件
UDI编码及其在GUDID数据库中的提交完整性
过往检查报告(尤其是483表和警告信)
医疗器械报告(MDR)数据库中的不良事件记录
召回、纠正和移除的相关信息
? 此外,FDA自2025年6月起在全中心部署了Elsa生成式AI平台,该平台可自动分析不良事件趋势、合规异常和历史检查结果,帮助检查员在出发前就锁定高风险领域。
⚠️ 对企业意味着:如果不提前自查注册列名有效性、UDI数据一致性、MDR提交记录等基础信息,现场检查时连“解释”的机会都很有限。
? 第二部分|新检查框架下的核心关注域 2.1 从QSIT到QMSR:检查逻辑的深层变化
? 在2026年2月之前,FDA长期使用“质量体系检查技术”(QSIT)作为检查方法,该方法将质量体系拆分为管理控制、设计控制、生产与过程控制、纠正预防措施(CAPA)等若干子系统,自上而下逐项核对。
? 随着QMSR的生效和CP 7382.850的启用,这一方法被全面替换。新程序不再沿用QSIT的子系统切割方式,而是要求检查员基于风险和数据驱动的思路,跨系统、跨流程地评估整个质量管理体系的有效性。QMSR正式纳入了ISO 13485:2016作为核心要求,同时引用ISO 9000:2015统一术语。
? 检查的核心问题从“你有没有这个文件”变成了“你的体系在实际运行中是否保持一致、能否形成闭环”。
2.2 内部审计与管理评审:一个容易被低估的变化
? 在过去二十多年的QSIT框架下,FDA检查员通常不会要求查看内部审计报告、管理评审记录或详细的供应商审计报告。这并非法律明文禁止,而是基于FDA内部政策形成的长期惯例——检查员可以确认企业“已完成内审/管理评审”的证据,但不能审阅报告的具体内容。
⚡ QMSR彻底改变了这一局面。通过纳入ISO 13485:2016的第8.2.2条(内部审计)和第5.6条(管理评审),FDA首次从法规层面明确授予检查员审查以下文件的权限:
? 内部审计的完整报告及支持性文档
? 管理评审会议的记录与决议
? 供应商审计的记录
? 相关培训文档
? 这意味着,企业不能再将内部审计和管理评审视为“内部私密文件”而拒绝提供。检查员还会将审计发现与本次检查结果进行比对,以验证内部审计程序是否真正有效。
2.3 投诉、MDR与CAPA的联动审查
? 上市后监督是FDA当前的重点关注领域。根据公开数据,2023年10月至2025年9月期间,FDA共开展了约4,400次医疗器械检查,其中38%被归类为上市后监督活动。
? 检查员在检查前就会调取企业的MDR数据。现场检查时,他们通常采用“样本追溯法”:随机选取几条投诉记录,然后逐项追问——
该投诉是否经过了充分调查?
调查结论是否触发了MDR判断?是否按规定时限提交了MDR?
如果启动了CAPA,纠正措施是否延伸到设计控制、生产过程或风险管理?
变更是否经过了验证?是否更新了相关文件?
❌ 未按规定提交MDR的行为,将被直接列入483表。 更重要的是,这条追溯路径实质上是在检验:企业的质量体系是否具备真正的闭环能力,而不仅仅是处理一个个孤立的客诉。
2.4 UDI与GUDID:数据一致性正成为执法重点
?️ UDI法规的实施采取分阶段合规日期的策略,不同类别的器械截止时间不同。对于已经到期的类别,UDI是强制性要求。
⚠️ FDA的执法行动正越来越多地聚焦于数据质量。现场检查中,检查员会直接核实物上的UDI编码与GUDID数据库中的记录是否一致——包括器械标识符(DI)、包装层级标识、生产标识符(如适用)以及当前状态。
? 一个UDI数据差异,足以构成483观察项。 此前已有企业因UDI标签不合规或GUDID信息不完整而收到警告信。
2.5 483表:从“恐惧”到“理性应对”
? FDA在检查结束时如果发现重大不符合项,会出具FDA 483表(检查观察报告)。这是一个事实陈述,而不是最终处罚。
⏱️ 企业在收到483表后,通常有15个工作日提交书面回应。部分检查员也允许企业在现场对483表的观察项进行批注,例如标注“已纠正并验证”“承诺纠正”等。但需要明确的是,现场批注并不是企业的强制性权利——是否接受、如何接受,取决于检查员的现场判断。
?️ 如果能在检查员离场前完成某些问题的整改并获得认可,确实有助于降低后续升级为警告信的风险。但最终是否发出警告信,取决于企业提交的正式整改回复是否能从体系层面解决根本问题。
? 值得注意的是,FDA的警告信数量正在上升。截至2025年9月初,针对医疗器械质量体系违规发出的警告信已达19封,超过了2024年同期总和。
?️ 第三部分|企业可以提前构建的四项能力 ✅ 能力一:把“证据链思维”嵌入日常运行
不要等到检查通知来了再去“补记录”。真正有效的准备,是在日常运行中就让每一条客诉、每一次变更、每一个CAPA都能够被追溯:
客诉是否经过了闭环调查?
是否触发了MDR判断?判断过程是否有记录?
CAPA的原因分析是否反推到设计或过程?
验证结果是否及时归档?
? 这套证据链不需要刻意“为检查而做”,但它必须是完整、可查阅、经得起抽样的。
✅ 能力二:以CP 7382.850为蓝本开展模拟演练
? 企业可以按照新合规程序手册的逻辑,定期组织内部模拟检查。模拟不应只停留在“看文件”,而要覆盖:
文件调取的速度与准确性
现场陪检人员的应答规范
跨部门信息协同的顺畅度
对检查员追问的应对能力
✅ 能力三:将内部审计和管理评审升级为“合规预检”
? 既然这些文件已经进入可检查范围,企业就不应再将其视为内部保密文件而敷衍了事。管理评审应当真实反映质量体系的运行状况,内部审计应当覆盖关键风险区域,审计发现与整改措施必须形成闭环记录。
✏️ 一个实用的做法:在每次管理评审或内部审计结束后,问自己一个问题——“如果检查员现在看到这份记录,他会认为我们的体系是有效的,还是有明显漏洞?”
✅ 能力四:提前完成UDI与GUDID的全量核查
? 建议企业对所有已到合规日期的产品,逐一比对:
实物标签上的UDI编码
包装层级的UDI编码
GUDID数据库中对应的记录
⚙️ 确保三端完全一致。同时,确认海外代理人具备足够的专业能力,能够在必要时协助FDA安排检查、传递信息,而不仅仅是一个“信箱”角色。
? 写在最后
FDA的检查方法从QSIT转向CP 7382.850,不是一次简单的版本更新,而是一次监管思维的系统性升级——从“查文件”走向“查系统”,从“看单点”走向“看闭环”。
? 能够从容应对这种检查的企业,不是那些临时抱佛脚、突击补记录的团队,而是那些在日常运营中已经把质量证据链做得清晰、完整、可追溯的团队。
? 合规从来不是一个部门的战斗,更不是一次检查的输赢。它是企业进入美国市场最基本的入场券,也是长期竞争中最扎实的护城河。
免责声明:本文内容仅供信息参考。FDA法规和检查要求可能随时更新,具体合规事宜请以FDA官方最新发布为准。
信息来源:网络新闻
排版整理:金飞鹰药械
✅ 本文基于FDA公开法规与检查程序手册整理,聚焦2026年QMSR生效背景下的检查要点与企业实操准备。
? 写在前面:为什么这篇文章值得读
FDA对医疗器械制造商的现场检查,是很多企业进入美国市场的“必过关卡”。但长期以来,行业讨论往往集中在“483表开了几条”这样的结果指标上,却很少有人系统拆解检查员背后的工作逻辑。
? 2026年2月,FDA正式完成了一次影响深远的法规切换:质量体系法规(QSR)被质量管理体系法规(QMSR)取代,同时启用全新的检查合规程序手册CP 7382.850。这意味着,检查员的关注点、取证方式、判断标准都与过去有了实质性不同。
? 本文试图回答三个问题:
FDA检查员在进场之前、现场之中、离场之后,分别做什么?
哪些环节最容易成为检查突破口?
企业如何提前构建一套能“自我证明”的质量体系?
? 第一部分|检查从来不是从进门开始的 FDA检查员的准备工作,远比大多数企业想象的更早、更深。
? 在确定检查计划后,检查员会通过FDA内部数据库和公开渠道,提前收集以下信息:
企业的注册信息和产品列名状态
510(k)或PMA的批准记录及任何补充文件
UDI编码及其在GUDID数据库中的提交完整性
过往检查报告(尤其是483表和警告信)
医疗器械报告(MDR)数据库中的不良事件记录
召回、纠正和移除的相关信息
? 此外,FDA自2025年6月起在全中心部署了Elsa生成式AI平台,该平台可自动分析不良事件趋势、合规异常和历史检查结果,帮助检查员在出发前就锁定高风险领域。
⚠️ 对企业意味着:如果不提前自查注册列名有效性、UDI数据一致性、MDR提交记录等基础信息,现场检查时连“解释”的机会都很有限。
? 第二部分|新检查框架下的核心关注域 2.1 从QSIT到QMSR:检查逻辑的深层变化
? 在2026年2月之前,FDA长期使用“质量体系检查技术”(QSIT)作为检查方法,该方法将质量体系拆分为管理控制、设计控制、生产与过程控制、纠正预防措施(CAPA)等若干子系统,自上而下逐项核对。
? 随着QMSR的生效和CP 7382.850的启用,这一方法被全面替换。新程序不再沿用QSIT的子系统切割方式,而是要求检查员基于风险和数据驱动的思路,跨系统、跨流程地评估整个质量管理体系的有效性。QMSR正式纳入了ISO 13485:2016作为核心要求,同时引用ISO 9000:2015统一术语。
? 检查的核心问题从“你有没有这个文件”变成了“你的体系在实际运行中是否保持一致、能否形成闭环”。
2.2 内部审计与管理评审:一个容易被低估的变化
? 在过去二十多年的QSIT框架下,FDA检查员通常不会要求查看内部审计报告、管理评审记录或详细的供应商审计报告。这并非法律明文禁止,而是基于FDA内部政策形成的长期惯例——检查员可以确认企业“已完成内审/管理评审”的证据,但不能审阅报告的具体内容。
⚡ QMSR彻底改变了这一局面。通过纳入ISO 13485:2016的第8.2.2条(内部审计)和第5.6条(管理评审),FDA首次从法规层面明确授予检查员审查以下文件的权限:
? 内部审计的完整报告及支持性文档
? 管理评审会议的记录与决议
? 供应商审计的记录
? 相关培训文档
? 这意味着,企业不能再将内部审计和管理评审视为“内部私密文件”而拒绝提供。检查员还会将审计发现与本次检查结果进行比对,以验证内部审计程序是否真正有效。
2.3 投诉、MDR与CAPA的联动审查
? 上市后监督是FDA当前的重点关注领域。根据公开数据,2023年10月至2025年9月期间,FDA共开展了约4,400次医疗器械检查,其中38%被归类为上市后监督活动。
? 检查员在检查前就会调取企业的MDR数据。现场检查时,他们通常采用“样本追溯法”:随机选取几条投诉记录,然后逐项追问——
该投诉是否经过了充分调查?
调查结论是否触发了MDR判断?是否按规定时限提交了MDR?
如果启动了CAPA,纠正措施是否延伸到设计控制、生产过程或风险管理?
变更是否经过了验证?是否更新了相关文件?
❌ 未按规定提交MDR的行为,将被直接列入483表。 更重要的是,这条追溯路径实质上是在检验:企业的质量体系是否具备真正的闭环能力,而不仅仅是处理一个个孤立的客诉。
2.4 UDI与GUDID:数据一致性正成为执法重点
?️ UDI法规的实施采取分阶段合规日期的策略,不同类别的器械截止时间不同。对于已经到期的类别,UDI是强制性要求。
⚠️ FDA的执法行动正越来越多地聚焦于数据质量。现场检查中,检查员会直接核实物上的UDI编码与GUDID数据库中的记录是否一致——包括器械标识符(DI)、包装层级标识、生产标识符(如适用)以及当前状态。
? 一个UDI数据差异,足以构成483观察项。 此前已有企业因UDI标签不合规或GUDID信息不完整而收到警告信。
2.5 483表:从“恐惧”到“理性应对”
? FDA在检查结束时如果发现重大不符合项,会出具FDA 483表(检查观察报告)。这是一个事实陈述,而不是最终处罚。
⏱️ 企业在收到483表后,通常有15个工作日提交书面回应。部分检查员也允许企业在现场对483表的观察项进行批注,例如标注“已纠正并验证”“承诺纠正”等。但需要明确的是,现场批注并不是企业的强制性权利——是否接受、如何接受,取决于检查员的现场判断。
?️ 如果能在检查员离场前完成某些问题的整改并获得认可,确实有助于降低后续升级为警告信的风险。但最终是否发出警告信,取决于企业提交的正式整改回复是否能从体系层面解决根本问题。
? 值得注意的是,FDA的警告信数量正在上升。截至2025年9月初,针对医疗器械质量体系违规发出的警告信已达19封,超过了2024年同期总和。
?️ 第三部分|企业可以提前构建的四项能力 ✅ 能力一:把“证据链思维”嵌入日常运行
不要等到检查通知来了再去“补记录”。真正有效的准备,是在日常运行中就让每一条客诉、每一次变更、每一个CAPA都能够被追溯:
客诉是否经过了闭环调查?
是否触发了MDR判断?判断过程是否有记录?
CAPA的原因分析是否反推到设计或过程?
验证结果是否及时归档?
? 这套证据链不需要刻意“为检查而做”,但它必须是完整、可查阅、经得起抽样的。
✅ 能力二:以CP 7382.850为蓝本开展模拟演练
? 企业可以按照新合规程序手册的逻辑,定期组织内部模拟检查。模拟不应只停留在“看文件”,而要覆盖:
文件调取的速度与准确性
现场陪检人员的应答规范
跨部门信息协同的顺畅度
对检查员追问的应对能力
✅ 能力三:将内部审计和管理评审升级为“合规预检”
? 既然这些文件已经进入可检查范围,企业就不应再将其视为内部保密文件而敷衍了事。管理评审应当真实反映质量体系的运行状况,内部审计应当覆盖关键风险区域,审计发现与整改措施必须形成闭环记录。
✏️ 一个实用的做法:在每次管理评审或内部审计结束后,问自己一个问题——“如果检查员现在看到这份记录,他会认为我们的体系是有效的,还是有明显漏洞?”
✅ 能力四:提前完成UDI与GUDID的全量核查
? 建议企业对所有已到合规日期的产品,逐一比对:
实物标签上的UDI编码
包装层级的UDI编码
GUDID数据库中对应的记录
⚙️ 确保三端完全一致。同时,确认海外代理人具备足够的专业能力,能够在必要时协助FDA安排检查、传递信息,而不仅仅是一个“信箱”角色。
? 写在最后
FDA的检查方法从QSIT转向CP 7382.850,不是一次简单的版本更新,而是一次监管思维的系统性升级——从“查文件”走向“查系统”,从“看单点”走向“看闭环”。
? 能够从容应对这种检查的企业,不是那些临时抱佛脚、突击补记录的团队,而是那些在日常运营中已经把质量证据链做得清晰、完整、可追溯的团队。
? 合规从来不是一个部门的战斗,更不是一次检查的输赢。它是企业进入美国市场最基本的入场券,也是长期竞争中最扎实的护城河。
免责声明:本文内容仅供信息参考。FDA法规和检查要求可能随时更新,具体合规事宜请以FDA官方最新发布为准。
信息来源:网络新闻
✅ 本文基于FDA公开法规与检查程序手册整理,聚焦2026年QMSR生效背景下的检查要点与企业实操准备。
? 写在前面:为什么这篇文章值得读
FDA对医疗器械制造商的现场检查,是很多企业进入美国市场的“必过关卡”。但长期以来,行业讨论往往集中在“483表开了几条”这样的结果指标上,却很少有人系统拆解检查员背后的工作逻辑。
? 2026年2月,FDA正式完成了一次影响深远的法规切换:质量体系法规(QSR)被质量管理体系法规(QMSR)取代,同时启用全新的检查合规程序手册CP 7382.850。这意味着,检查员的关注点、取证方式、判断标准都与过去有了实质性不同。
? 本文试图回答三个问题:
FDA检查员在进场之前、现场之中、离场之后,分别做什么?
哪些环节最容易成为检查突破口?
企业如何提前构建一套能“自我证明”的质量体系?
FDA检查员的准备工作,远比大多数企业想象的更早、更深。
? 在确定检查计划后,检查员会通过FDA内部数据库和公开渠道,提前收集以下信息:
企业的注册信息和产品列名状态
510(k)或PMA的批准记录及任何补充文件
UDI编码及其在GUDID数据库中的提交完整性
过往检查报告(尤其是483表和警告信)
医疗器械报告(MDR)数据库中的不良事件记录
召回、纠正和移除的相关信息
? 此外,FDA自2025年6月起在全中心部署了Elsa生成式AI平台,该平台可自动分析不良事件趋势、合规异常和历史检查结果,帮助检查员在出发前就锁定高风险领域。
⚠️ 对企业意味着:如果不提前自查注册列名有效性、UDI数据一致性、MDR提交记录等基础信息,现场检查时连“解释”的机会都很有限。
2.1 从QSIT到QMSR:检查逻辑的深层变化
? 在2026年2月之前,FDA长期使用“质量体系检查技术”(QSIT)作为检查方法,该方法将质量体系拆分为管理控制、设计控制、生产与过程控制、纠正预防措施(CAPA)等若干子系统,自上而下逐项核对。
? 随着QMSR的生效和CP 7382.850的启用,这一方法被全面替换。新程序不再沿用QSIT的子系统切割方式,而是要求检查员基于风险和数据驱动的思路,跨系统、跨流程地评估整个质量管理体系的有效性。QMSR正式纳入了ISO 13485:2016作为核心要求,同时引用ISO 9000:2015统一术语。
? 检查的核心问题从“你有没有这个文件”变成了“你的体系在实际运行中是否保持一致、能否形成闭环”。
2.2 内部审计与管理评审:一个容易被低估的变化
? 在过去二十多年的QSIT框架下,FDA检查员通常不会要求查看内部审计报告、管理评审记录或详细的供应商审计报告。这并非法律明文禁止,而是基于FDA内部政策形成的长期惯例——检查员可以确认企业“已完成内审/管理评审”的证据,但不能审阅报告的具体内容。
⚡ QMSR彻底改变了这一局面。通过纳入ISO 13485:2016的第8.2.2条(内部审计)和第5.6条(管理评审),FDA首次从法规层面明确授予检查员审查以下文件的权限:
? 内部审计的完整报告及支持性文档
? 管理评审会议的记录与决议
? 供应商审计的记录
? 相关培训文档
? 这意味着,企业不能再将内部审计和管理评审视为“内部私密文件”而拒绝提供。检查员还会将审计发现与本次检查结果进行比对,以验证内部审计程序是否真正有效。
2.3 投诉、MDR与CAPA的联动审查
? 上市后监督是FDA当前的重点关注领域。根据公开数据,2023年10月至2025年9月期间,FDA共开展了约4,400次医疗器械检查,其中38%被归类为上市后监督活动。
? 检查员在检查前就会调取企业的MDR数据。现场检查时,他们通常采用“样本追溯法”:随机选取几条投诉记录,然后逐项追问——
该投诉是否经过了充分调查?
调查结论是否触发了MDR判断?是否按规定时限提交了MDR?
如果启动了CAPA,纠正措施是否延伸到设计控制、生产过程或风险管理?
变更是否经过了验证?是否更新了相关文件?
❌ 未按规定提交MDR的行为,将被直接列入483表。 更重要的是,这条追溯路径实质上是在检验:企业的质量体系是否具备真正的闭环能力,而不仅仅是处理一个个孤立的客诉。
2.4 UDI与GUDID:数据一致性正成为执法重点
?️ UDI法规的实施采取分阶段合规日期的策略,不同类别的器械截止时间不同。对于已经到期的类别,UDI是强制性要求。
⚠️ FDA的执法行动正越来越多地聚焦于数据质量。现场检查中,检查员会直接核实物上的UDI编码与GUDID数据库中的记录是否一致——包括器械标识符(DI)、包装层级标识、生产标识符(如适用)以及当前状态。
? 一个UDI数据差异,足以构成483观察项。 此前已有企业因UDI标签不合规或GUDID信息不完整而收到警告信。
2.5 483表:从“恐惧”到“理性应对”
? FDA在检查结束时如果发现重大不符合项,会出具FDA 483表(检查观察报告)。这是一个事实陈述,而不是最终处罚。
⏱️ 企业在收到483表后,通常有15个工作日提交书面回应。部分检查员也允许企业在现场对483表的观察项进行批注,例如标注“已纠正并验证”“承诺纠正”等。但需要明确的是,现场批注并不是企业的强制性权利——是否接受、如何接受,取决于检查员的现场判断。
?️ 如果能在检查员离场前完成某些问题的整改并获得认可,确实有助于降低后续升级为警告信的风险。但最终是否发出警告信,取决于企业提交的正式整改回复是否能从体系层面解决根本问题。
? 值得注意的是,FDA的警告信数量正在上升。截至2025年9月初,针对医疗器械质量体系违规发出的警告信已达19封,超过了2024年同期总和。
✅ 能力一:把“证据链思维”嵌入日常运行
不要等到检查通知来了再去“补记录”。真正有效的准备,是在日常运行中就让每一条客诉、每一次变更、每一个CAPA都能够被追溯:
客诉是否经过了闭环调查?
是否触发了MDR判断?判断过程是否有记录?
CAPA的原因分析是否反推到设计或过程?
验证结果是否及时归档?
? 这套证据链不需要刻意“为检查而做”,但它必须是完整、可查阅、经得起抽样的。
✅ 能力二:以CP 7382.850为蓝本开展模拟演练
? 企业可以按照新合规程序手册的逻辑,定期组织内部模拟检查。模拟不应只停留在“看文件”,而要覆盖:
文件调取的速度与准确性
现场陪检人员的应答规范
跨部门信息协同的顺畅度
对检查员追问的应对能力
✅ 能力三:将内部审计和管理评审升级为“合规预检”
? 既然这些文件已经进入可检查范围,企业就不应再将其视为内部保密文件而敷衍了事。管理评审应当真实反映质量体系的运行状况,内部审计应当覆盖关键风险区域,审计发现与整改措施必须形成闭环记录。
✏️ 一个实用的做法:在每次管理评审或内部审计结束后,问自己一个问题——“如果检查员现在看到这份记录,他会认为我们的体系是有效的,还是有明显漏洞?”
✅ 能力四:提前完成UDI与GUDID的全量核查
? 建议企业对所有已到合规日期的产品,逐一比对:
实物标签上的UDI编码
包装层级的UDI编码
GUDID数据库中对应的记录
⚙️ 确保三端完全一致。同时,确认海外代理人具备足够的专业能力,能够在必要时协助FDA安排检查、传递信息,而不仅仅是一个“信箱”角色。
? 写在最后
FDA的检查方法从QSIT转向CP 7382.850,不是一次简单的版本更新,而是一次监管思维的系统性升级——从“查文件”走向“查系统”,从“看单点”走向“看闭环”。
? 能够从容应对这种检查的企业,不是那些临时抱佛脚、突击补记录的团队,而是那些在日常运营中已经把质量证据链做得清晰、完整、可追溯的团队。
? 合规从来不是一个部门的战斗,更不是一次检查的输赢。它是企业进入美国市场最基本的入场券,也是长期竞争中最扎实的护城河。
免责声明:本文内容仅供信息参考。FDA法规和检查要求可能随时更新,具体合规事宜请以FDA官方最新发布为准。
信息来源:网络新闻
排版整理:金飞鹰药械

往期精彩推荐
往期精彩推荐
医疗器械注册咨询认准金飞鹰
深圳:0755-86194173
广州:020 - 82177679
湖南:0731-22881823
湖北:181-3873-5940
江苏:135-5494-7827
广西:188-2288-8311
海南:135-3810-3052
重庆:135-0283-7139

