
当前,国内大量 中小微企业(SMEs) 在面对欧盟 MDR 与 IVDR 新法规时,最怕陷入“合规成本黑洞”与“发证遥遥无期”的生死局。但真实的权威数据显示,欧盟市场 90% 的认证基本盘全部是 SME。法规从底层逻辑上并没有赶尽杀绝,反而留下了合法的“豁免通道”:合规负责人(PRRC)允许外包以削减人力成本,公告机构(NB)被强制要求提供 SME 专属减免费率,官方甚至鼓励免费的“结构性对话”来降低试错风险。看懂法规底牌,停止盲目烧钱,善用豁免特权与精简产品线,才是中小企业在 2025 年监管重压下逆风翻盘的唯一出路。
做欧盟医疗器械合规咨询这些年,我们听过国内企业抱怨最多的一句话就是:“我们几十个人的小厂,哪有财力养那么庞大的法规团队去搞 MDR 甚至 IVDR?”
确实,从旧指令(MDD/IVDD)升级到医疗器械法规(Regulation (EU) 2017/745, MDR)和体外诊断医疗器械法规(Regulation (EU) 2017/746, IVDR),监管门槛呈指数级上升,临床评价、上市后监督等要求给中小微企业带来了极其沉重的资金压力和行政负担。
很多企业甚至产生了严重的“合规幻觉”,认为欧洲市场以后就是跨国巨头的专属游乐场。但事实真的如此吗?今天,我们将跳出枯燥的教条,结合欧盟公告机构(Notified Body, NB)近三年的真实行业调查数据,为您深度拆解:中小企业如何利用法规赋予的“法定特权”,合法、合规地实现降本增效。
认清基本盘:你不是弱势群体,欧盟监管的“基本盘”正是 SMEs
在探讨如何省钱之前,我们必须先打破一个信息差:公告机构(NB)并没有抛弃中小企业。
纵观欧盟官方在 2023 年至 2025 年底发布的历次 NB 机构行业调查报告,我们可以清晰地看到一个极度集中的趋势:中小企业(SMEs)在欧洲医疗器械和体外诊断市场中占据了绝对主导地位。
在最新的大规模权威样本调查中,数据令人震撼:
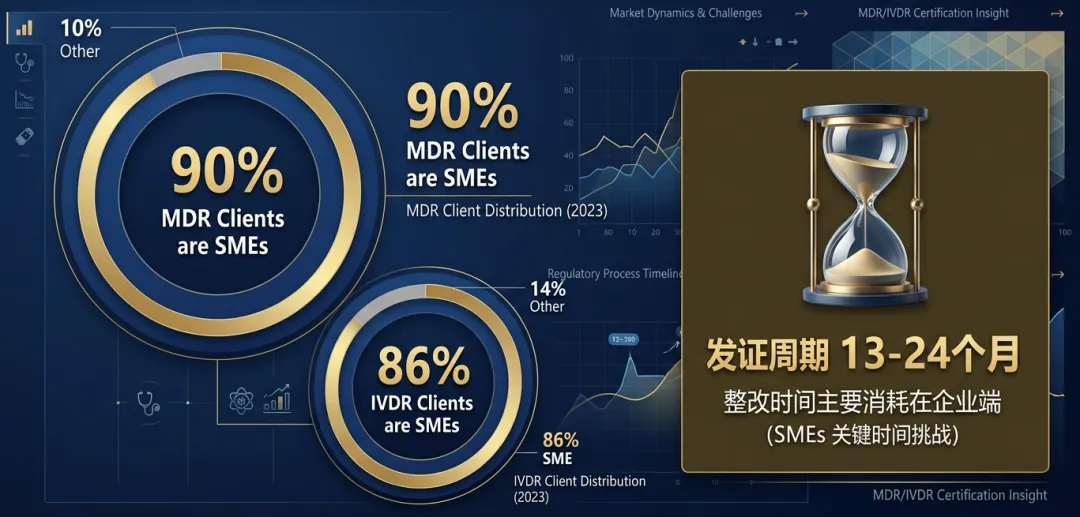
MDR 总盘:在总计超过一万三千家 MDR 客户中,90% 为中小企业(SMEs),大型企业仅占 10%。更夸张的是,高达 73% 的 NB 机构明确表示,其手头超过 90% 的客户全部都是中小企业。 IVDR 总盘:在 IVDR 客户中,86% 为中小企业(SMEs),超过一半的 IVDR 机构表示其绝大部分客户是 SME。 出海军团画像:无论是 MDR 还是 IVDR,非欧盟客户的占比均超过一半,而这些来自中国、美国等地的制造商,同样以 SME 为主。
既然 NB 机构的日常审核工作完全是围绕 SME 展开的,为什么国内 SME 依然感觉“拿证难如登天”?
报告揭示了 SME 认证失败和周期无限拉长的两个核心痛点:
第一是“申请被拒(Application not complete)”和“超出机构业务范围”。因为缺乏庞大专业的法规注册团队,很多 SME 在没有吃透 MDCG 指南和产品分类代码的前提下盲目提交,导致初审直接被退回。
第二是“整改耗时”。如今获取一张完整的 MDR/IVDR 证书(包含质量体系与产品)平均需要 13 到 24 个月,而其中超过一半的时间,是消耗在制造商自身对不符合项(NCs)的整改和补充数据上。时间成本正在透支 SME 的现金流。
NB survey
因此,
SME 破局的关键,不在于拼财力,
而在于拼“合规策略的精确度”。
降本第一刀:合法外包合规负责人(PRRC),巧用“微小企业豁免权”
在人力成本方面,MDR 与 IVDR 第 15 条(Article 15)引入了一个让无数中小企业头疼的核心岗位:合规负责人(Person responsible for regulatory compliance, PRRC)。
法规对 PRRC 的硬性门槛极高:要么具备相关科学学位并外加至少 1 年的专业经验,要么直接具备 4 年的专业合规经验。对于地处中国内陆的中小微医疗器械厂来说,招募这样一位精通欧盟法规的双语高端人才,不仅年薪高昂,且极难留住。
但是,法规在这里给 SME 开了一个极其重要的合法“后门”。
实操建议:合规外包(Outsourcing) 法规明确规定,符合欧盟 2003/361/EC 建议定义的微型和小型企业(Micro and small enterprises),不需要在组织内部全职雇佣 PRRC。企业只需确保该负责人“永久且持续地(permanently and continuously at their disposal)”为其提供服务即可。
如何判定你是否属于微型和小型企业? 根据欧盟定义,员工人数少于 50 人,且年营业额或资产总额不超过 1000 万欧元的企业,即可享受此项豁免。
MDR/IVDR
企业可以合法地将 PRRC 的职责分包给第三方专业咨询机构或外部独立专家。但在实操中,您必须通过严谨的商业动作来规避合规风险:
建立实质性合同连接:不能仅仅口头约定,必须与外部专家签订正式的服务合同。 资质证明入档:合同附件中必须包含该外部专家的学位证书、履历证明,以自证其满足法规的学历和经验硬性要求。 响应机制设计:既然是“永久且持续”,专家无需 24 小时坐在你的办公室,但合同必须规定其在产品放行审批、技术文档签署、以及发生严重不良事件时的警戒报告(Vigilance)环节,具备明确的介入时效和处理权限。
通过这一机制,微小企业可以把数百万的全职人力成本,转化为可控的外包服务费,极大减轻了运营负担。
拿捏机构软肋:理直气壮申请 SME 专属费率与排期倾斜
很多 SME 在向公告机构(NB)询价时,看到动辄数十万甚至上百万人民币的审核账单,往往直接倒吸一口凉气,认为这是死规定,只能咬牙接受。实际上,你完全有法定权利要求更合理的报价。
实操建议:主动亮明 SME 身份 为了确保欧洲内部市场的公平性并避免阻碍创新,MDR/IVDR 附件 VII(Annex VII)针对 NB 机构设定了明确的义务:NB 机构在制定标准费用清单和服务条款时,必须是公平合理的,并且必须明确考虑中小企业(SME)的利益。
此外,在最新的行业标准操作指南中,NB 机构在发布报价前,强制要求先获取制造商的基本信息,其中必须包含“是否为中小企业(SME)”这一关键尽调指标。
因此,SME 在预申请(Pre-application)阶段,绝不能仅仅提交产品资料,必须采取以下策略:
提交身份证明:主动、正式地向 NB 机构提交员工人数、年营业额等财务与人事信息,证明自身符合欧盟 2003/361/EC 关于 SME 的定义。 要求费用透明:在比价和合同谈判环节,理直气壮地要求 NB 机构说明其报价明细中,是否以及如何体现了对 SME 的减免或特殊考量。在某些特定程序(例如高风险器械的临床评价磋商程序)中,SME 依法甚至可以获得法定费用的直接减免。 争取产能排期:MDCG(医疗器械协调小组)多次呼吁各方建立机制,为 SME 和首次申请者分配审核产能。遇到 NB 机构以“产能排满”为由推诿时,可以合法援引相关政策导向,要求合理的排期资源倾斜。
激活 - 结构性对话
(Structured Dialogue)
我们在调查报告中看到,导致 SME 认证失败的首要原因是“申请材料不完整”和“超出机构业务范围”。很多国内 SME 习惯了闭门造车,拿着旧版技术文档直接向 NB 递交,结果换来几十个不符合项(NCs),不仅消耗了宝贵的过渡期缓冲时间,每次整改重审还在无形中增加了高昂的审核时效费。
实操建议:用好官方免费沟通渠道 法规虽然严禁 NB 机构提供任何关于“如何实现合规”的具体咨询服务,但为了避免资源浪费,MDCG 强烈鼓励公告机构与制造商在符合性评估的申请前或申请中,组织“结构性对话(Structured Dialogues)”。
这是一种为了交流技术信息和法规指导的合法机制,它不违反 NB 的独立性与公正性,更不应被视为需要额外收费的咨询项目。鉴于试错成本极高,聪明的 SME 已经开始将其作为合规战略的“标准起手式”。
在正式砸下重金开展大规模临床评估或体系升级前,SME 应主动发起结构性对话,重点确认以下问题:
产品分类规则对齐:确认您的产品在 MDR/IVDR 下的分类是否发生跳级,以及对应的 NB 机构代码(MDN/MDA等)是否在其授权范围内。 遗留设备(Legacy Devices)路径:讨论旧产品技术文件的分批提交计划。 抽样与评估路径:确认具体需要满足哪些标准(what needs to be fulfilled),明晰评估路径。
通过这种前置性的边界确认,SME 能够精准投放有限的资源,彻底避免方向跑偏。
生死时速:死守质量管理体系(QMS)与过渡期底线
得益于欧盟 (EU) 2023/607 号法规(针对 MDR)和 (EU) 2024/1860 号法规(针对 IVDR)的修订,原本即将到期的遗留设备过渡期获得了大幅度延长。
例如在 IVDR 方面,D 类设备的过渡期延至 2027 年底,C 类延至 2028 年底,B 类和 A 类无菌设备延至 2029 年底。但如果中小企业因此认为可以“彻底躺平”,那就犯了致命错误。
要享受这些诱人的延期,法规设定了极其严苛的“占坑”前提,其中最核心的底线就是质量管理体系(Quality management system, QMS)的时间节点。
实操建议:先搭骨架,再填血肉 对于 MDR,企业必须在 2024 年 5 月 26 日之前建立符合第 10(9) 条要求的 QMS,并提交正式申请。 对于 IVDR,SME 必须牢记以下红线节点:
必须在 2025 年 5 月 26 日之前,建立符合 IVDR 第 10(8) 条要求的质量管理体系(QMS)。 必须在规定日期前(D 类 2025 年 5 月,C 类 2026 年 5 月等)向 NB 机构提交合格评定申请(Formal application)。 必须在提交申请后四个月内签署书面协议(Written agreement)。
对于资源有限的 SME 而言,务实的 QMS 升级策略是怎样的? 公告机构在过渡期早期,并不强求您的体系能够立刻产出完美的临床评价报告(CER)或复杂的风险管理数据。但是,您的 QMS 必须已经充分融入了新法规中关于上市后监督(Post-market surveillance, PMS)、警戒系统(Vigilance)、经济运营商(Economic operators)注册等核心监管要求。
这意味着,如果您的体系依然是停留在十年前 ISO 13485 旧框架下的“空壳体系”,错过了上述的底线日期,您的产品将直接丧失过渡期资格,面临强制退市的灾难性后果。
活命哲学:断尾求生与警惕新规“断供”暗雷
在历届的 NB 调查中,我们发现一个残酷的现象:很多 SME 申请失败或撤回的原因,是“经济原因或未准备好”,以及“无法解决不符合项”。
由于 MDR/IVDR 极大地拉高了临床证据(如 PMCF/PMPF)和上市后监督(PMS)的门槛,维持一个老旧产品合规所需的维护成本,甚至可能超过了该产品一年的净利润。
实操建议一:大刀阔斧的产品线精简(Portfolio Rationalization) 中小企业必须摒弃“产品越全越好”的旧思维。未来 2-3 年内,活下去的唯一方式是进行果断的产品线精简。企业必须对现有目录进行全盘财务核算,对于那些低利润、低附加值、或者历史临床数据极其薄弱的老旧产品,必须要有“断臂求生”的勇气,果断砍掉。集中有限的现金流和法规资源,全力保住核心拳头产品的欧盟准入资格。
实操建议二:防范 IVDR 第 10a 条的断供预警暗雷 对于 IVD 企业,近期还有一个极易踩坑的监管盲区。中小企业由于现金流紧张、供应链相对脆弱,更容易遭遇原材料断供或产能危机。
根据最新生效(自 2025 年 1 月 10 日起)的 IVDR 第 10a 条(Article 10a),如果制造商预计某种关键设备的供应将中断,且这种中断可能对患者或公共卫生造成严重伤害,制造商必须提前履行通知义务。
除非遭遇不可抗力的突发天灾,否则该中断预警通知必须在预计发生前至少 6 个月提交给主管当局、相关经济运营商(Economic operators)以及医疗机构。国内 SME 必须立刻将这一规定写入自身的 QMS 警戒与上市后监督流程中,否则一旦因资金链断裂导致停产,不仅丢了市场,还会引来欧盟主管当局的巨额合规重罚。
结语:
精细化运营是 SME 跨越合规鸿沟的唯一跳板
欧盟 MDR 和 IVDR 绝不是只有跨国巨头才能玩得转的权力游戏,它的本质依然是一个由中小企业(SMEs)撑起的浩瀚大市场。
对于国内出海的医疗器械 SME 而言,合规之路已经从“拼运气、拼财力”的粗放时代,全面转向了“拼合规策略、拼理解力”的排位赛。准确界定微小企业身份落实 PRRC 外包、毫不妥协地要求 NB 机构提供 SME 专属减免费率、主动发起结构性对话锁定评估路径、并死守QMS 的生死线,才是把每一分钱都花在刀刃上的生存法则。
未来的欧洲市场竞争法则非常残酷:谁的法规团队更具备实操智慧、谁的技术文档能够做到一次性通过、谁敢于果断砍掉吸血的边缘产品,谁就能在这场属于中小企业的合规大逃杀中脱颖而出。看懂了规则,看似严苛的法规,反而会成为你甩开竞争对手、建立长期壁垒的护城河。
