
摘要: 随着 COVID-19 大流行期间 mRNA 疫苗技术的成功商业化,合同研发与生产组织(CDMO)在全球疫苗供应链中的作用变得至关重要。本文系统性地介绍了在高速 mRNA 灌装生产线调试与确认(CQV)过程中应用的质量与合规策略。
1. 背景与挑战
1.1 行业背景
后疫情时代,全球对 mRNA 疫苗的需求持续增长。负责任的制造商在加速推进产能扩张的同时,需要继续从现有产能中提供救命疫苗。引入最先进的灌装线以保障疫苗供应链安全,已成为应对当前和未来大流行的关键任务。
1.2 新生产线建设的关键目标
mRNA 生产线调试与确认的核心目标包括建设以下功能区域:
| 洁净区域 | |
| 处理区域 | |
| 核心生产区 | |
| 支持系统 | |
| 质控区域 | |
| 公用工程 |
1.3 设计重点
设备与设施设计需聚焦于:
自动化水平:实现高度自动化和数据监控 生物安全:满足二级生物安全水平(BSL-2) 技术整合:将患者安全、质量和生产力导向的新技术融入现有质量体系
2. 设定标准与规划阶段
2.1 工程规划阶段的关键活动
在现有场地采用尖端技术涉及复杂的工程规划,包括:
调试与确认重新配置、安装、建设与退役规划
评估变更对现有质量管理体系的影响 开发各类技术规范文档
2.2 关键文档体系
2.3 质量部门早期全面参与的必要性
与"减少质量参与"的普遍认知相反,此类项目需要质量部门及产品/工艺主题专家(SME)的全面前期参与,原因如下:
1) 确定业务适配性
从业务发展团队获取潜在项目类型、需求技术和灵活解决方案的输入 确保在产品未明确时具备所需灵活性
2) 初始质量风险管理(QRM)
与 ASTM 标准一致的风险识别应用 基于识别风险开发 CQV 计划 不完整的风险评估可能导致必要测试用例被排除
3) 指导 URS 开发
将 URS 作为即时里程碑并不理想 建立敏捷且平衡的质量-SME-供应商团队更为关键 通过头脑风暴会议构建知识库
4) 规范开发
需考虑产品/工艺要求和监管趋势 特别关注制造操作数据完整性和数据治理
2.4 适用的行业指南
表1:关键行业指南清单
| PDA | |
| ISPE | |
| ASTM | |
| ICH |
2.5 ASTM E2500 的混合应用
[[ASTM E2500-20-制药系统规范设计与验证标准指南|ASTM E2500]] 强调应用风险管理和质量源于设计(QbD)理念,确保关键产品/工艺属性内建于设备/系统中。然而,对于 CDMO 而言,产品/工艺信息并非总能预先获得用于设备试运行,因此混合方法更为理想:
采用适合计划产能的技术 避免重复测试 支持基于科学和风险的决策制定
3. 风险管理策略
3.1 基于风险的方法
ASTM 和 ISPE 均推荐采用基于风险的方法驱动活动。[[ICH Q9-质量风险管理|ICH Q9]] 质量风险管理指南提倡:
使用基于科学的风险评估保护患者安全 确保风险管理努力程度与风险水平相称
3.2 风险评估工具选择
早期阶段(数据有限时)
流程图和地图 鱼骨图(石川图) 检查表 故障树分析 失效模式与影响分析([[FMEA-失效模式与影响分析|FMEA]]/[[FMECA-失效模式影响与危害度分析|FMECA]]) 初步危害分析 危害可操作性分析([[HAZOP-危害与可操作性分析|HAZOP]]) 危害分析与关键控制点([[HACCP-危害分析与关键控制点|HACCP]])
数据充足后
系统性的、数据驱动的风险评估工具
3.3 风险评估生命周期
根据可用增量新数据,在以下 CQV 阶段执行风险评估: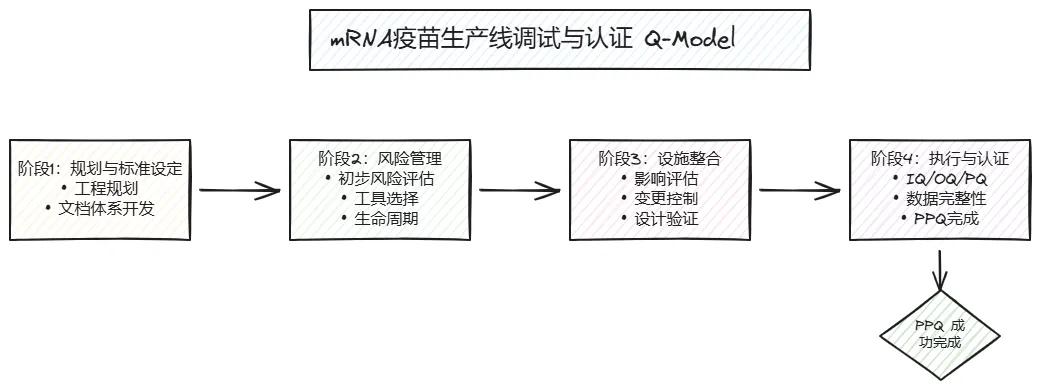
概念阶段:基于文献、供应商知识和可用信息 设计阶段:基于设计规范和功能规范 调试阶段:基于设备测试和运行数据 认证阶段:基于性能确认数据
3.4 ICH Q9 修订的新要求
[[ICH Q9-质量风险管理|ICH Q9]] 草案修订要求决策者最小化风险评估的主观性。使用设计不良的风险评分工具可能引入主观性,因此需要在 CQV 每个阶段证明所使用风险评估工具的合理性。
4. 现有设施整合考量
4.1 影响评估要点
对于已有运营的设施,需评估新增生产线或设备的影响:
表2:整合影响评估维度
| 公用工程 | |
| 人员 | |
| 物料流 | |
| 检测资源 | |
| 程序文件 | |
| 施工影响 | |
| 环境监控 | |
| 报警管理 |
4.2 变更控制的应用
[变更控制]是记录影响和跟踪缓解措施的有效工具:
根据施工进度和项目计划可能需要临时措施和修订 质量部门可独立评估对质量管理体系的影响 在施工开始前解决差距以支持顺利执行
4.3 设计验证文档
包含现有设施、设备和公用工程的设计验证文档是良好实践,用于验证:
每个相关系统均已审查 系统已证明能够满足工艺要求
5. 执行、审查、批准与利用
5.1 数据完整性的重要性
CQV 活动及其产生的数据具有永久性,将在设施整个生命周期内的检查中被检索和审查。
⚠️ 关键数据:
FDA 2012-2019 年警告信审查显示,**55%**的问题与数据完整性相关 其中 45% 的数据完整性观察与生产操作数据相关
应对措施:
持续培训良好文件规范([[GDP-良好文件规范|GDP]]) 强化数据治理 特别关注涉及多个 CQV 供应商的快速项目
5.2 责任明确与利用策略
CQV 计划需明确执行责任方。[[ASTM E2500-20-制药系统规范设计与验证标准指南]] 支持基于风险的认证,允许通过利用调试来支持认证状态。
表3:利用决策检查清单
5.3 质量部门早期参与
强烈建议在调试阶段早期涉及质量部门:
设计测试计划 修改任何测试 管理潜在测试失败
这确保质量部门以促进利用决策和最大限度减少测试案例重复的方式同意调试方法。
6. 项目成功标准
6.1 成功定义
CQV 项目只有在制造操作完美满足以下要求时才视为成功:
美国 FDA 工艺验证指南 EU GMP 附件15 其他相关监管指南
[[PPQ-工艺性能确认]] 是关键里程碑。
6.2 团队解散时机
理想情况下,只有在成功完成 PPQ 后才应解散 CQV 项目团队。传达 CQV 项目成功将确保质量部门、主题专家和供应商的早期参与。
6.3 额外下游工作评估
应开发矩阵计算和评估由于缺乏 CQV 数据收集所需额外下游工作的成本,包括:
确定初始设置运行参数 确定操作范围 确定自动化控制参数
7. 结论与最佳实践
7.1 理想执行策略
快速项目的理想 CQV 执行阶段需要:
质量部门现场支持:初步文件审查与执行同步进行 协议开发时机:设备供应商知识转移完成后起草 试运行后开发:安排在供应商现场或现场试运行后开发协议 一次做对([[RTF-Right First Time]]):将 RTF 矩阵应用于协议
7.2 质量评估
执行协议或总结报告的质量应基于审查过程中观察到的以下错误数量评估:
GDP 错误 技术错误 数据完整性错误
7.3 关键成功因素
表4:CQV 项目关键成功因素
| 绩效矩阵 | |
| 头脑风暴 | |
| 持续沟通 | |
| 资源规划 | |
| KPI 体系 |
8. 参考文献
Xie, W, Chen, B, Wong, J. "Evolution of the market for mRNA technology," Nature Reviews/Drug Discovery 2021. [[ASTM E2500-20-制药系统规范设计与验证标准指南|ASTM E2500-20]]: Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment FDA Guidance for Industry: Process Validation: General Principles and Practices [[ICH Q9-质量风险管理|ICH Q9]]: Quality Risk Management ISPE Baseline Guide Vol. 5: Commissioning and Qualification PDA TR 54-5: Quality Risk Management for the Design, Qualification, and Operation of Manufacturing Systems PDA TR 84: Integrating Data Integrity Requirements into Manufacturing & Packaging Operations Pazhayattil et al. "A Semi-Quantitative Risk Assessment Methodology Fit for Bio-Pharmaceutical Lifecycle Stages," PDA J Pharm Sci Tech 2020
关于作者
Ahmed Elsaid - Emergent Biosolution 洛克维尔分公司质量高级总监,在注射剂制造领域拥有超过21年质量领导经验。Ajay Babu Pazhayattil - Capcium Inc. 科学和监管事务副总裁,在制药和生物制药运营的各领域成功实现可衡量成果。
本文综合了 PDA 发布的《A Successful Q-Model for New mRNA Line Commissioning & Qualification》Part 1 和 Part 2 的内容。
