近日,我院内分泌科室收治一例尿液外观呈红葡萄酒样的老年女性患者。如此少见的尿液颜色引起了我们的注意,该患者尿液颜色如此奇怪,究竟是何原因导致,让我们一起解开谜团。
患者,女,65岁,以“发现血糖升高15年余,酱油色尿10余天”为主诉,收治入院。15年前体检发现血糖升高,无口干、口渴,无多饮、多尿,不伴体重下降,按“糖尿病”给予降糖治疗,降糖方案“二甲双胍、格列吉特缓释片、阿卡波糖”,平素未检测血糖。曾于我院住院治疗,出院诊断“2型糖尿病、糖尿病周围神经病变、糖尿病伴大血管病变”。10余天前无明显诱因出现酱油色尿,自行多饮水后逐渐缓解,3天前上述症状再发,伴发热、畏寒、恶心。于当地医院检查:尿常规隐血阳性、尿蛋白阳性,为进一步诊疗,就诊于门诊。高血压病史7年,最高血压180/90mmHg,目前应用“硝苯地平缓释片”降压治疗,血压控制尚可。既往慢性乙肝病史,病程不详。无冠心病、结核史,无重大手术、外伤史,无药物及食物过敏史,预防接种史随当地。

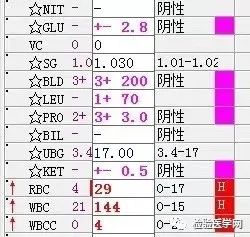
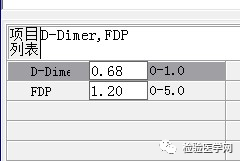
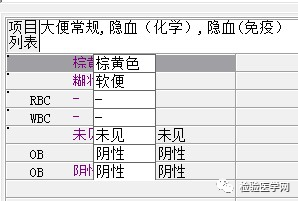
尿红白细胞易见,隐血强阳性、蛋白阳性,尿糖、酮体阳性。结合病史,考虑慢性病引起的泌尿系统感染及肾功能受损导致的酱油色血尿?尿隐血强阳性,是血红蛋白尿?肌红蛋白尿?依次为贫血五项、巨幼贫两项、凝血六项、粪便隐血、血常规、生化常规、尿常规、尿红细胞形态。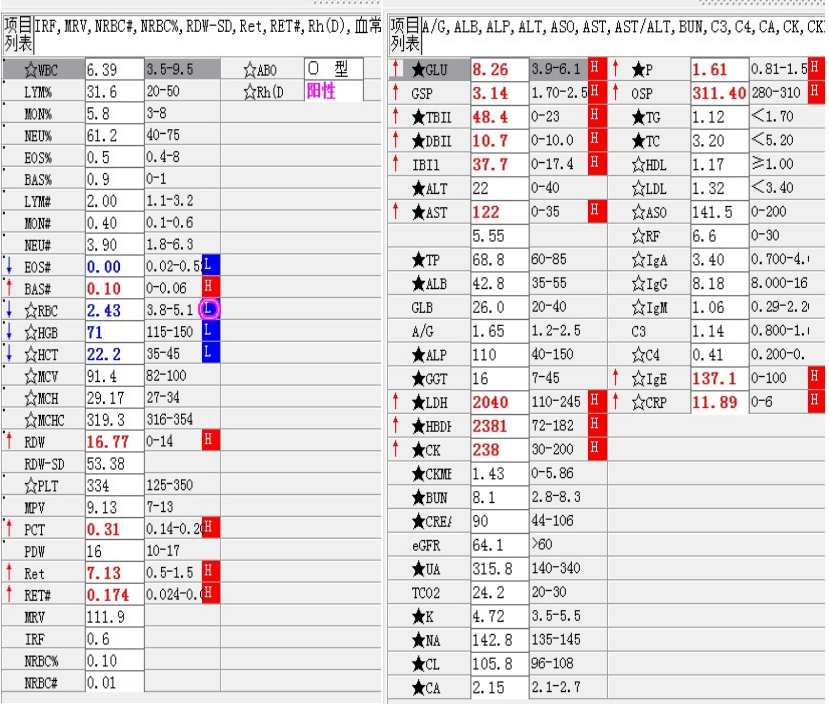
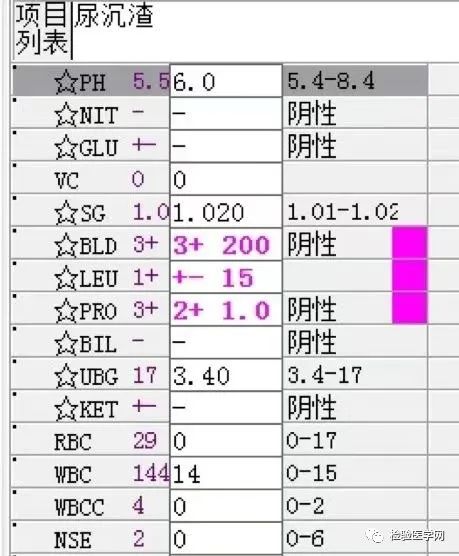
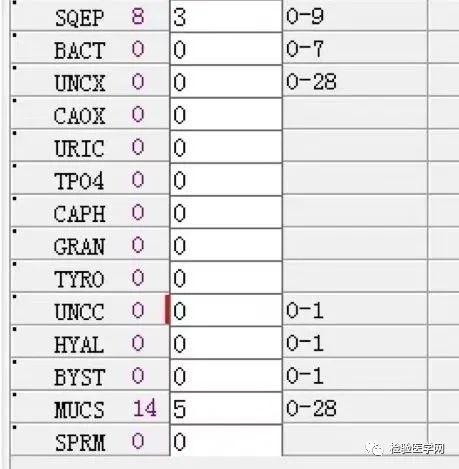
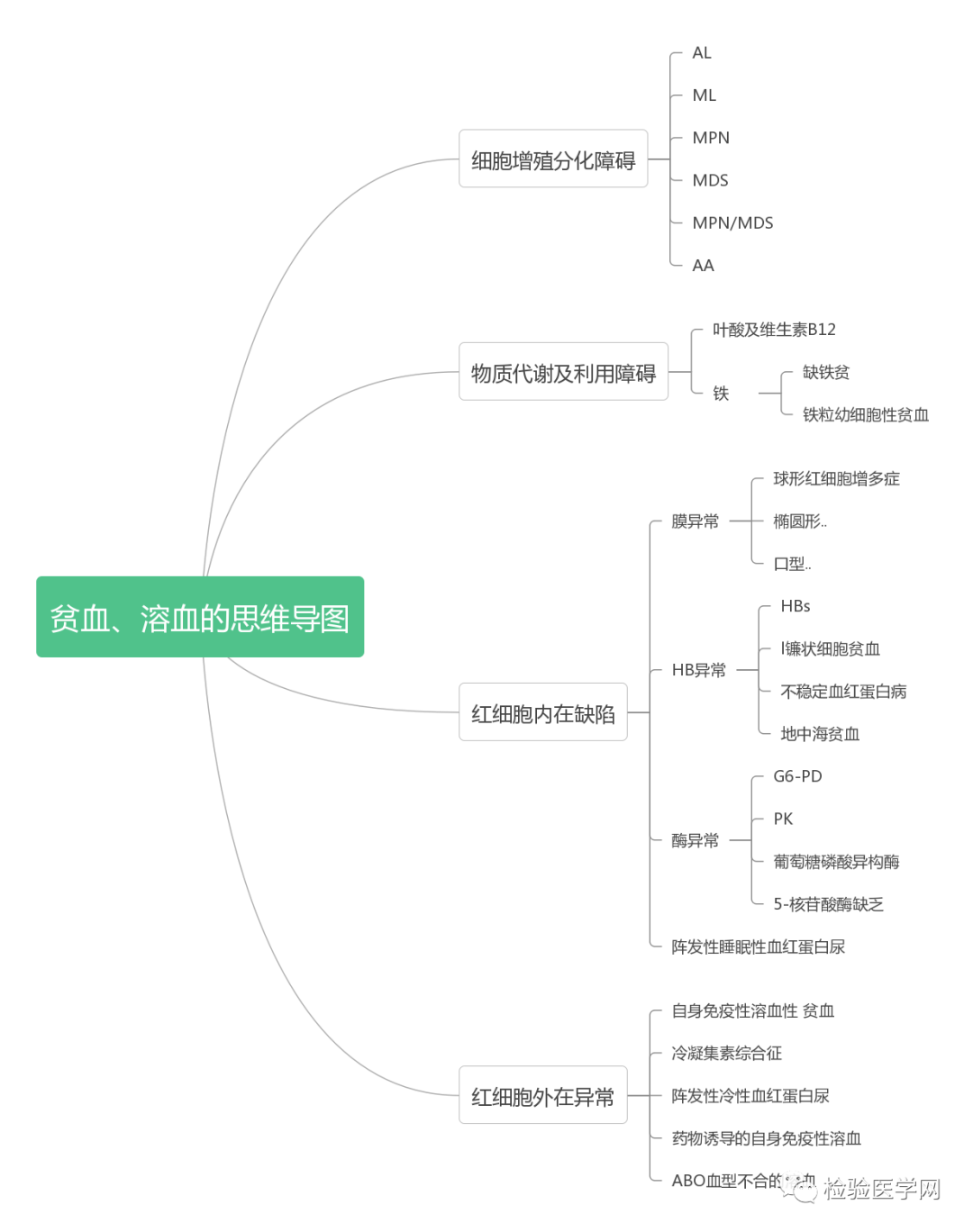
1. MCV、MCH、MCHC正常,缺铁五项正常,巨幼两项正常,排除最常见的缺铁性贫血和巨幼贫。2. 红细胞数量明显减低,中度贫血,网织红细胞明显增高,考虑红细胞寿命缩短或者病理性破坏增加、骨髓代偿能力不足导致的贫血。3. 生化游离胆红素、乳酸脱氢酶、肌酸激酶等的增加,符合溶血特征。4. 血糖、谷草、糖化白蛋白增高,符合糖尿病、乙肝携带者病史。IgE增高,待分析。5.尿液酱油色,尿红细胞数量正常,且形态未见明显异常,排除肾小球损害,考虑血红蛋白尿、血尿后红细胞溶解、肌红蛋白尿等。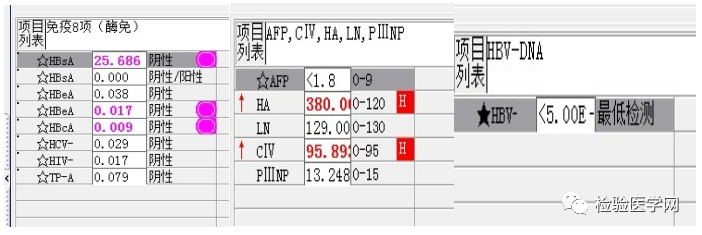
乙肝小三阳,肝纤维化指标异常,但乙肝病毒载量未检测到,建议高敏测定乙肝病毒。
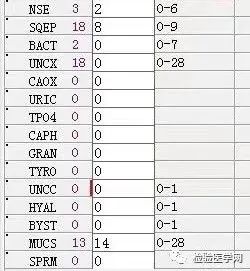
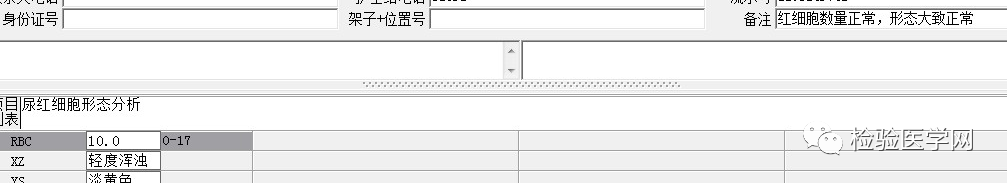
尿本周蛋白阴性,尿红细胞形态正常,考虑排除糖尿病引起的肾功能损害。尿含铁血红素实验阳性、尿隐血强阳性,提示血管内溶血;应对溶血性疾病进行分析:多见于遗传性溶血性贫血、获得性溶血性贫血、阵发性血红蛋白尿,行军性血红蛋白尿等。不规则抗体阴性、抗核抗体阴性、免疫球蛋白正常,考虑排除自身免疫性疾病引起的溶血。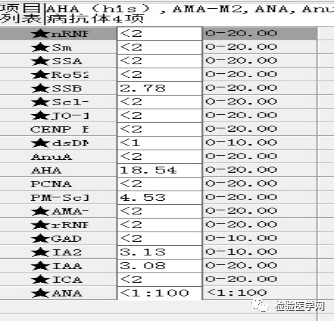
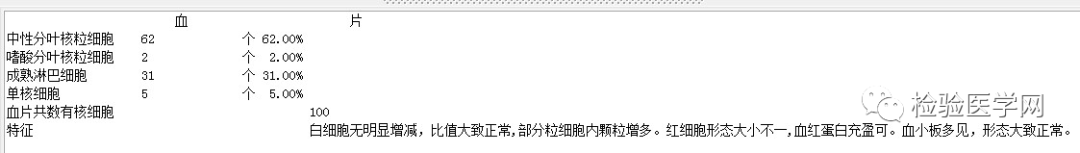

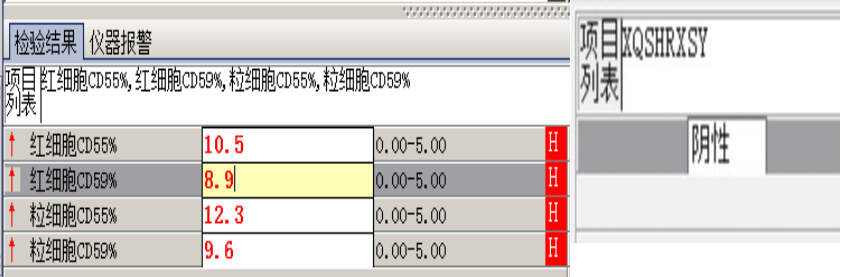
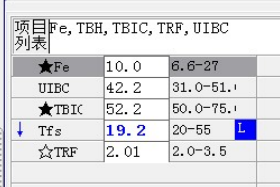
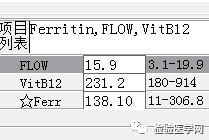
外周血形态仅见红细胞大小不一,未见明显异常红细胞形态。骨髓未见明显幼稚细胞,红系增生活跃,符合骨髓代偿情况。酸化血清溶血试验:阴性。CD55/59结果:阴性表达占比增高。肝胆胰脾彩超:肝实质回声粗糙,肝囊肿,胆囊内透声差。甲状腺彩超:甲状腺右叶多发混合性结节,甲状腺双叶多发囊性结节。心脏彩超:右房增大,二、三尖瓣及主动脉瓣少量反流,左室舒张功能减低。颈部及下肢血管彩超;双侧绞颈总动脉斑块形成,右侧椎动脉斑块形成,双下肢动脉硬化。胡桃夹综合彩超:未见明显“胡桃夹”声像图表现。胸片示:双肺纹理增强;主动脉硬化。心电图:示正常小电图。1. 肾功三项、尿红细胞形态、尿本周蛋白正常,提示肾脏功能大致正常,排除重吸收减弱及肾HB阈值改变引起的血红蛋白尿。肝功能异常,间接胆红素、LDH、HBDH增加,符合溶血特点。2. 复检尿常规,红白细胞阴性,尿隐血依然强阳性,心肌酶谱仅CK增高,结合病史,排除肌红蛋白尿,考虑血红蛋白尿病因。3. 铁三项、五项排除缺铁贫;叶酸维B排除巨幼贫;外周血红细胞形态可以排除细胞膜缺陷引起的贫血(多为遗传性,比如球形红细胞、靶形红细胞、口型红细胞等,易见红细胞碎片增多);自身抗体Coombs实验阴性,排除自身免疫性溶贫;骨髓象分析未见明显原幼细胞、病态造血排除AA,排除恶性血液病。4. 酸化血清溶血试验阳性,Rous试验阳性,CD55、CD59低表达细胞群占比增高,综合分析,考虑诊断为阵发性睡眠性血红蛋白尿。【溶血性贫血的实验室诊断思路】
最终诊断:阵发性睡眠性血红蛋白尿。
阵发性睡眠性血红蛋白尿是一种由于一个或多个造血干细胞Xp22.1上PIG-A基因突变导致的获得性造血干细胞克隆性疾病,其发病机制为PIG-A基因突变导致全血细胞(干细胞及其所有子代细胞)膜糖化磷脂酰肌醇(GPI)锚合成障碍,造成血细胞表面GPI锚链接蛋白缺失(CD55和CD59),细胞灭火补体能力减弱,从而引起细胞溶血。基因突变——锚链蛋白异常——细胞膜蛋白55、59缺失——减少补体抵抗——溶血;补体异常激活对血细胞、干细胞损伤——骨髓衰竭。临床表现为疲劳、嗜睡、虚弱、腹部疼痛、胸疼、呼吸困难等,多因为不同程度的发作性血管内溶血、平滑肌功能障碍、造血功能衰竭、静脉血栓形成等。PNH的血栓形成倾向,其机制尚未明确,常发生于不同寻常的部位,多见肝静脉血栓,其次为肠系膜、脑静脉、下肢深静脉血栓形成。扩展:在其他血液学疾病的情况下,PNH亦有一定概率并发。建议增加免疫抑制治疗。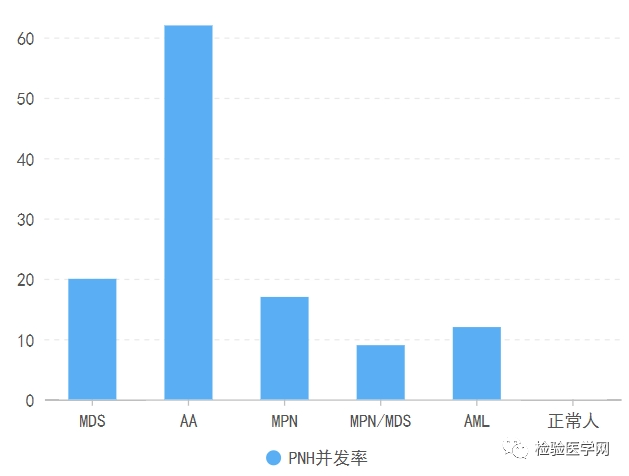
疑问:该患者为什么仅一系减少?怀疑乙肝肝病的影响使珠蛋白合成减少,血管内溶血又导致HB含量增加,超出珠蛋白结合能力以及肾阈值,进而提前出现大量血红蛋白尿。血细胞减少,常见三系减少;铁减少,长期尿铁丢失过多;血管内溶血(LDH、HBDH、间接胆红素上升,珠蛋白减低);流失细胞术(CD55/CD59阴性细胞大于10%,FLAER阴性细胞占比>1%);尿含铁血黄素定性试验阳性;酸化血清溶血试验阳性;PIGA基因缺陷。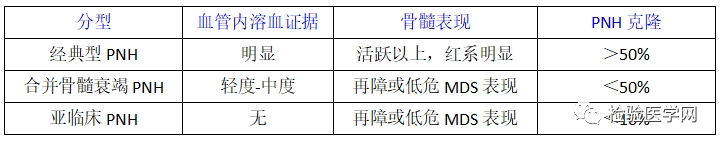
临床表现分级:①贫血分级:极重度 HGB≤30g/L、重度 HGB 31~60g/L、中度 HGB 61-90g/L、轻度 HGB>90 g/L;②血红蛋白尿分级:频发 ≤2个月发作 1次、偶发 >2个月发作1次、不发 观察2年无发作(观察不足2年未发为暂不发)。Ham试验、糖水试验、蛇毒因子溶血试验、尿潜血(或尿含铁血黄素)等项试验中凡符合下述任何一种情况,即可诊断:①2项以上阳性;②1项阳性,但须具备下列条件:a. 2次以上阳性,或1次阳性,但操作正规、有阴性对照、结果可靠,即时重复仍阳性者;b.有溶血的其他直接或间接证据,或有肯定的血红蛋白尿出现;c.能除外其他溶血,特别是遗传性球形红细胞增多症、自身免疫性溶血性贫血、葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症所致的溶血和阵发性冷性血红蛋白尿症等。流式细胞术检测发现,外周血中CD55或CD59阴性,中性粒细胞或红细胞>10%(5%~10%为可疑)。
临床表现符合,实验室检查具备1项或2项者皆可诊断,1、2两项可以相互佐证。凡AA转化为PNH,或同时兼有两病特征而以某病为主,可将本综合征再分为四种情况:1. AA-PNH:指原有肯定的AA(或未能诊断的PNH早期表现),转化为确定的PNH,AA的表现已不明显。
2. PNH-AA:指原有肯定的PNH(而非下述的第4类),转为明确的AA,PNH的表现已不明显。
3. PNH伴有AA特征:指临床及实验室检查所见均说明病情仍以PNH为主,但伴有1个或1个以上部位骨髓增生低下、有核细胞减少、网织红细胞不增高等AA表现者。
4. AA伴有PNH特征:指临床及实验室检查所见均说明病情仍以AA为主,但具有PNH的实验室诊断结果阳性者。
国际PNH工作组(I-PIG)将PNH患者分为如下几类:经典型PNH:该类患者有典型的溶血和血栓形成;合并其他骨髓衰竭性疾病:如AA或MDS;亚临床型PNH:患者有微量PNH克隆,但没有溶血和血栓的实验室和临床证据。PNH的传统治疗手段仍然是以“保护”PNH克隆、减少补体攻击和破坏,减轻溶血为目的,以对症支持治疗为主,如无禁忌证,在急性溶血发作时,可予肾上腺糖皮质激素(如泼尼松 0.25-1.00mg·kg~·d ,为避免长期应用的不良反应,应酌情短周期使用),辅以细胞膜稳定剂(维生素E)、叶酸及碱性药物(如碳酸氢钠)治疗,对多数初诊患者能减轻溶血发作、稳定病情。为防止加重溶血,不常规给予铁剂。若为PNH-AA综合征可辅助雄激素(如十一酸睾酮、达那唑)、免疫抑制剂(如环孢素)等治疗。PNH患者是否采取血栓的抗凝预防,目前尚无定论。对于发生血栓者应给予抗凝和肝素治疗。其他对症支持治疗包括必要时输注红细胞、血小板以及出现感染时给予抗菌药物。(1) 重组人源型抗补体蛋白c5单克隆抗体(Eeulizum-ab,Soliris):Eeulizumab是抑制末端补体成分活化的重组人源型单克隆抗体(单抗),能特异性地结合到人末端补体蛋白c5,通过抑制人补体c5向C5a和C5b的裂解以阻断炎症因子 C5a的释放及C5b-9的形成。研究表明,该抗体对c5有高度亲和力,能阻断C5a和C5b-9的形成,并保护哺乳动物细胞不受C5b-9介导的损伤。由于该单抗抑制机体的免疫系统功能,从而增加了患者对某些严重感染的易感性。国外报道用药期间易出现细菌性脑膜炎。临床试验证实Eeulizumab治疗PNH可显著减轻血管内溶血,减少红细胞输注,明显改善PNH患者贫血,减少血栓形成,延长生存期。Eeulizumab于2007年3月16日被美国FDA批准用于治疗PNH,推荐剂量每周静脉滴注600mg,用4次,第5周900mg,以后每2周900mg,持续12周。(2) 联合化疗:对于激素原发耐药、继发耐药或激素依赖的溶血不易控制、反复发作的骨髓增生良好的PNH患者,为有效地减少PNH异常克隆,最大限度地控制溶血,可采用化疗,利用正常克隆较PNH克隆耐受补体能力强,对造血生长因子反应好,正常造血恢复快的优势,使正常克隆逐步取代PNH克隆,而达到治疗目的。可采用减低剂量的DA(柔红霉素+阿糖胞苷)或HA(高三尖杉酯碱+阿糖胞苷)方案之后,加造血刺激因子(G-CSF和EPO)。实践证明,化疗能够有效地减少PNH克隆负荷、控制溶血、改善贫血,而且大大减少了激素的用量,是一种较有应用前景的治疗手段。但为避免出现化疗后骨髓抑制期的严重并发症(贫血、出血和严重感染),化疗采用的剂量应偏小,疗程亦应缩短;应加强隔离和保护,预防感染;应重用造血因子促进正常克隆恢复。3. 异基因造血干细胞移植(allo-HSCT)在补体蛋白C5单抗应用之前,allo-HSCT治疗一般限于那些难治性、耐肾上腺皮质激素或有激素禁忌征的PNH患者,适应征为:有HLA相合的同胞供者,且满足以下条件:①合并骨髓衰竭;②难治性PNH,输血依赖性溶血性贫血;③反复出现危及生命的血栓栓塞事件。目前,上述情况均可通过补体蛋白C5单抗得以全部或部分控制,故最合适的移植适应征目前仍无定论。PNH患者妊娠期间病死率较高,治疗主要以输注红细胞和血小板,改善贫血和预防出血为主。目前Eeulizumab尚未批准应用于妊娠期妇女,必要时给予低分子量肝素预防血栓直至婴儿出生后6周。但PNH的治疗仍很复杂,需要有经验的血液科医生和产科医生携手处理高风险的妊娠。